Вабисмо раствор д/ин. 120 мг/мл по 0.24 мл (28.8 мг) №1 во флак. с иголк.
Ф. Хоффманн-Ля Рош Лтд. (Швейцарія)
Состав
діюча речовина: faricimab;
1 флакон містить 28,8 мг фарицимабу в 0,24 мл розчину, що відповідає концентрації 120 мг/мл;
разова доза – 6 мг/0,05 мл;
допоміжні речовини: L-гістидин, кислота оцтова 30 %, L-метіонін, натрію хлорид, D-сахароза, полісорбат 20, вода для ін’єкцій.
Лікарська форма
Розчин для ін’єкцій.
Основні фізико-хімічні властивості: прозора або опалесцентна рідина, безбарвного або коричнювато-жовтого кольору.
Фармакотерапевтична група
Засоби, що діють на органи чуття. Засоби, що застосовуються в офтальмології. Засоби при судинних очних захворюваннях. Антинеоваскуляризаційні засоби. Фарицимаб.
Код АТХ S01L A09.
Фармакологічні властивості
Фармакодинаміка.
Механізм дії
Фарицимаб є гуманізованим біспецифічним антитілом IgG1, що діє через пригнічення двох окремих шляхів передачі сигналу за рахунок нейтралізації як Ang-2 (ангіопоетин-2), так і фактора росту ендотелію судин A (VEGF-A). Ang-2 спричиняє васкулярну нестабільність шляхом стимуляції дестабілізації ендотелію, втрати перицитів і патологічного ангіогенезу, потенціюючи таким чином випотівання рідини через судини і запалення. Ang-2 також сенсибілізує кровоносні судини до дії VEGF-A, що призводить до подальшої васкулярної дестабілізації. Ang-2 і VEGF-A синергічно збільшують проникність судин і стимулюють неоваскуляризацію. Шляхом подвійного пригнічення Аng-2 і VEGF-A фарицимаб зменшує проникність судин і запалення, інгібує патологічний ангіогенез і відновлює васкулярну стабільність.
У описаних нижче чотирьох клінічних дослідженнях III фази було виявлено супресію середньої концентрації вільного ANG-2 і вільного VEGF-A в органах зору, починаючи з дня 7, порівняно із вихідним рівнем.
Неоваскулярна (ексудативна) вікова макулярна дегенерація (нВМД)
Подібне при застосуванні афліберцепту зменшення середньої товщини центру фовеолярної зони (товщина центрального підполя, CST) спостерігалося, починаючи з вихідного рівня по 48 тиждень під час застосування препарату Вабісмо. Середнє зменшення CST від вихідного рівня до візитів для оцінки первинної кінцевої точки (усереднено на 40, 44 і 48 тижнях) становило -137 мкм і -137 мкм для препарату Вабісмо з інтервалом введення 8 тижнів (кожні 8 тижнів), 12 тижнів (кожні 12 тижнів) або 16 тижнів (кожні 16 тижнів) порівняно із -129 мкм і -131 мкм при застосуванні афліберцепту в дослідженнях TENAYA і LUCERNE відповідно. Tаке середнє зменшення CST підтримувалось впродовж року 2. Спостерігався порівнянний ефект препаратів Вабісмо і афліберцепту щодо зменшення інтраретинальної рідини (IRF), субретинальної рідини (SRF) і відшарування пігментного епітелію (ВПЕ). Під час візитів для оцінки первинної кінцевої точки в дослідженнях TENAYA і LUCERNE частка пацієнтів із відсутністю IRF становила відповідно 76–82 % і 78–85 % при лікуванні препаратом Вабісмо порівняно з 74–85 % та 78–84 % при лікуванні афліберцептом. Відсоток пацієнтів із відсутністю SRF у двох дослідженнях становив 70–79 % і 66–78 % при лікуванні препаратом Вабісмо порівняно з 66–78 % і 62–76 % при лікуванні афліберцептом. Відсоток пацієнтів із відсутністю ВПЕ у двох дослідженнях становив 3–8 % і 3–6 % при лікуванні препаратом Вабісмо порівняно з 8–10 % та 7–9 % при лікуванні афліберцептом. Таке зменшення IRF, SRF і ВПЕ підтримувалось впродовж року 2 (тижні 104–108).
На 48 тижні порівнянні зміни від вихідного рівня показника загальної площі ураження, спричиненого хоріоїдальною неоваскуляризацією (CNV), і порівнянне зменшення зони випотівання CNV з екскрецією крові та рідини спостерігалися в обох дослідженнях у пацієнтів, які отримували лікування препаратом Вабісмо та афліберцептом.
Діабетичний макулярний набряк (ДМН)
Зниження середнього значення CST від вихідного рівня, що спостерігалося в дослідженнях YOSEMITE і RHINE, було чисельно більшим у пацієнтів, які отримували лікування препаратом Вабісмо з інтервалом кожні 8 тижнів та з інтервалом до кожних 16 тижнів коригованого дозування, порівняно з тими, хто отримував афліберцепт кожні 8 тижнів у період з 4 по 100 тиждень. В обох дослідженнях у більшої частки пацієнтів у групах препарату Вабісмо з часом було досягнуто відсутності IRF і відсутності ДМН (визначено як досягнення CST менше 325 мкм за результатами оптичної когерентної томографії [ОКТ]) порівняно з групою афліберцепту. Порівнянне зменшення SRF з часом спостерігалося в групах лікування препаратами Вабісмо і афліберцептом в обох дослідженнях. Середнє зниження CST від вихідного рівня до візитів для оцінки первинної кінцевої точки (усереднено на 48, 52 і 56 тижнях) у дослідженні YOSEMITE становило 207 мкм і 197 мкм у пацієнтів, які отримували лікування препаратом Вабісмо з інтервалом кожні 8 тижнів та з інтервалом до кожних 16 тижнів коригованого дозування, порівняно зі 170 мкм у пацієнтів, які отримували лікування афліберцептом кожні 8 тижнів; у дослідженні RHINE результати становили 196 мкм, 188 мкм і 170 мкм відповідно. Таке зменшення середнього значення CST зберігалося до 2 року. Частка пацієнтів із відсутністю ДМН під час візитів для оцінки первинної кінцевої точки (мінімальне/максимальне значення) в дослідженні YOSEMITE становила 77–87 % і 80–82 % у групі лікування препаратом Вабісмо з інтервалом кожні 8 тижнів і з інтервалом до кожних 16 тижнів коригованого дозування відповідно порівняно з 64–71 % у групі лікування афліберцептом кожні 8 тижнів; у дослідженні RHINE результати становили 85–90 %, 83–87 % і 71–77 % відповідно. Ці результати зберігались до 2 року.
У дослідженні YOSEMITE частка пацієнтів із відсутністю IRF під час візитів для оцінки первинної кінцевої точки (усереднено на 48, 52 і 56 тижнях) становила 42–48 % і 34–43 % у групі лікування препаратом Вабісмо з інтервалом кожні 8 тижнів і з інтервалом до кожних 16 тижнів коригованого дозування відповідно порівняно з 22–25 % у групі лікування афліберцептом кожні 8 тижнів; у дослідженні RHINE результати становили 39–43 %, 33–41 % і 23–29 % відповідно. Ці результати зберігалися до 2 року.
Клінічна ефективність та безпека
Лікування нВМД
Ефективність та безпека застосування фарицимабу порівняно з анти-VEGF лікуванням вивчалися в двох рандомізованих (1:1) багатоцентрових подвійно сліпих дослідженнях з двома групами (TENAYA і LUCERNE) за участю пацієнтів із нВМД. Лікування (фарицимаб, 6 мг, або афліберцепт, 2 мг) проводилося шляхом інтравітреальної ін’єкції початково із 4-тижневими інтервалами. В групі афліберцепту після 3 початкових ін’єкцій інтервал між введеннями становив 8 тижнів до кінця дослідження (кожні 8 тижнів). У групі фарицимабу інтервал між введеннями було скориговано індивідуально після 4 початкових доз. Заключний (фіксований) інтервал між введеннями становив 8 тижнів (кожні 8 тижнів), 12 тижнів (кожні 12 тижнів) або максимум 16 тижнів (кожні 16 тижнів) залежно від встановленої протоколом зміни CST, визначеної за результатами спектральної оптичної когерентної томографії та/або зміни BCVA, визначеної за кількістю літер за дослідженням ефективності раннього лікування діабетичної ретинопатії (ETDRS), a також від клінічної оцінки лікарем наявності/відсутності крововиливу в жовту пляму на 20 і 24 тижні. Починаючи з тижня 60 і далі, пацієнти групи лікування Вабісмо були переведені на гнучкий режим дозування, відповідно до якого інтервал між введеннями можна було збільшувати з кроком до 4 тижнів (до кожних 16 тижнів) або можна було зменшувати з кроком до 8 тижнів (до кожних 8 тижнів) на основі автоматизованої об’єктивної оцінки попередньо визначених критеріїв візуальної та анатомічної активності захворювання. Пацієнти в групі афліберсепту продовжували отримувати режим дозування кожні 8 тижнів протягом усього періоду дослідження. Обидва дослідження тривали 112 тижнів.
Дослідження включали загалом 1329 раніше нелікованих пацієнтів, при цьому 1135 (85 %) пацієнтів завершили дослідження до тижня 112. Загалом 1326 пацієнтів отримали щонайменше одну дозу (включно з 664 пацієнтами групи фарицимабу). Середній вік [діапазон віку] досліджуваної популяції становив 75,9 року [від 50 до 99 років]. Первинною кінцевою точкою ефективності була середня зміна BCVA від вихідного рівня (на основі середнього значення протягом 40, 44 і 48 тижнів), визначена за допомогою таблиці літер ETDRS з відстані 4 метри. В обох дослідженнях первинна гіпотеза (не менша ефективність) була підтверджена: пацієнти, які отримували лікування препаратом Вабісмо з інтервалом до кожних 16 тижнів, і пацієнти, які отримували лікування афліберцептом кожні 8 тижнів, продемонстрували порівнянну середню зміну BCVA від відповідного вихідного показника через 1 рік. Значне покращення зору від вихідного стану відмічалось до тижня 112 в обох групах лікування. Детальні результати обох досліджень наведено в таблицях 1 і 2, а також на рисунку 1.
Частка пацієнтів в групах персоналізованих інтервалів введення препарату на 48 тижні в дослідженнях TENAYA і LUCERNE відповідно становила: кожні 16 тижнів – 46 %, 45 %; кожні 12 тижнів – 34 %, 33 %; кожні 8 тижнів – 20 %, 22 %.
Частка пацієнтів в групах персоналізованих інтервалів введення препарату на 112 тижні в дослідженнях TENAYA і LUCERNE відповідно становила: кожні 16 тижнів – 59 %, 67 %; кожні 12 тижнів – 15 %, 14 %; кожні 8 тижнів – 26 %, 19 %.
Tаблиця 1
Результати щодо ефективності під час візитів для оцінки первинної кінцевої точкиа через 2 рокиb в дослідженні TENAYA
|
Результати щодо ефективності |
TENAYA |
|||
|
Рік 1 |
Рік 2 |
|||
|
Вабісмо, інтервал введення до кожних 16 тижнів, N = 334 |
Афліберцепт кожні 8 тижнів, N = 337 |
Вабісмо, інтервал введення до кожних 16 тижнів, N = 334 |
Афліберцепт кожні 8 тижнів, N = 337 |
|
|
Середня зміна BCVA від вихідного показника за результатами визначення кількості літер за ETDRS (95 % ДІ) |
5,8 (4,6, 7,1) |
5,1 (3,9, 6,4) |
3,7 (2,1, 5,4) |
3,3 (1,7, 4,9) |
|
Частка пацієнтів зі збільшенням розпізнавання на ≥ 15 літер від вихідного рівня (зважена частка за CMH, 95 % ДІ) |
20 % (15,6 %, 24,4 %) |
15,7 % (11,9 %, 19,6 %) |
22,5 % (17,8 %, 27,2 %) |
16,9 % (12,7 %, 21,1 %) |
|
Частка пацієнтів, які уникнули зменшення розпізнання на ≥ 15 літер від вихідного рівня (зважена частка за CMH, 95 % ДІ) |
95,4 % (93 %, 97,7 %) |
94,1 % (91,5 %, 96,7 %) |
92,1 % (89,1 %, 95,1 %) |
88,6 % (85,1 %, 92,2 %) |
a Середнє тижнів 40, 44 і 48.
b Середнє тижнів 104, 108, 112.
BVCA – найкраща скоригована гострота зору; ETDRS – дослідження ефективності раннього лікування діабетичної ретинопатії; ДІ – довірчий інтервал; СMH – метод Кокрана – Мантеля – Хензеля, статистичний тест, який генерує оцінку зв’язку з бінарним результатом і використовується для оцінки категоріальних змінних.
Tаблиця 2
Результати щодо ефективності під час візитів для оцінки первинної кінцевої точкиа через 2 рокиb в дослідженні LUCERNE
|
Результати щодо ефек6тивності |
LUCERNE |
|||
|
Рік 1 |
Рік 2 |
|||
|
Вабісмо, інтервал введення до кожних 16 тижнів, N = 331 |
Афліберцепт кожні 8 тижнів, N = 327 |
Вабісмо, інтервал введення до кожних 16 тижнів, N = 331 |
Афліберцепт кожні 8 тижнів, N = 327 |
|
|
Середня зміна BCVA від вихідного показника за результатами визначення кількості літер за ETDRS (95 % ДІ) |
6,6 (5,3, 7,1) |
6,6 (5,3, 7,8) |
5,0 (3,4, 6,6) |
5,2 (3,6, 6,8) |
|
Частка пацієнтів зі збільшенням розпізнавання на ≥ 15 літер від вихідного рівня (зважена частка за CMH, 95 % ДІ) |
20,2 % (15,9 %, 24,6 %) |
22,2 % (17,7 %, 26,8 %) |
22,4 % (17,8 %, 27,1 %) |
21,3 % (16,8 %, 25,9 %) |
|
Частка пацієнтів, які уникнули зменшення розпізнання на ≥ 15 літер від вихідного рівня (зважена частка за CMH, 95 % ДІ) |
95,8 % (93,6 %, 98,0 %) |
97,3 % (95,5 %, 99,1 %) |
92,9 % (90,1 %, 95,8 %) |
93,2 % (90,2 %, 96,2 %) |
a Середнє тижнів 40, 44 і 48.
b Середнє тижнів 104, 108, 112.
BVCA – найкраща скоригована гострота зору; ETDRS – дослідження ефективності раннього лікування діабетичної ретинопатії; ДІ – довірчий інтервал; СMH – метод Кокрана – Мантеля – Хензеля, статистичний тест, який генерує оцінку зв’язку з бінарним результатом і використовується для оцінки категоріальних змінних.
|
Скоригована середня зміна від вихідного показника |
|
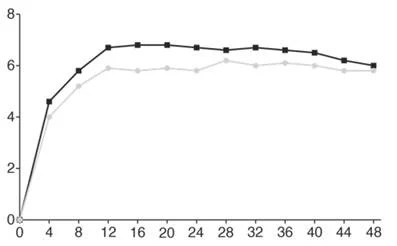
Тижні
 Фарицимаб, 6 мг, до кожні 16 тижнів (N = 665)
Фарицимаб, 6 мг, до кожні 16 тижнів (N = 665)
 Афліберцепт, 2 мг, кожні 8 тижнів (N = 664)
Афліберцепт, 2 мг, кожні 8 тижнів (N = 664)
Рис. 1.Об’єднані дослідження III фази нВМД (TENAYA і LUCERNE): графік зміни BCVA від вихідного рівня в досліджуваному оці до 112 тижня, метод MMRM (первинна оцінка) (популяція ITT)
В обох дослідженнях TENAYA і LUCERNE покращення BCVA та CST від вихідного рівня на 60 тижні було порівнянним у двох групах лікування і збігалося з тим, що спостерігалося на 48 тижні.
Результати щодо ефективності в усіх підгрупах пацієнтів, що оцінювалися (наприклад, за віком, статтю, етнічним походженням, вихідною гостротою зору, типом ураження, розміром ураження) в кожному дослідженні та в сукупному аналізі, збігалися з результатами в загальній популяції.
В обох дослідженнях препарат Вабісмо, який вводили з інтервалом до кожних 16 тижнів, продемонстрував клінічно значуще покращення від вихідного рівня до 48 тижня сумарного індексу за опитувальником для оцінки зорової функції Національного офтальмологічного інституту США (NEI VFQ-25), що був порівнянним із таким при застосуванні афліберцепту кожні 8 тижнів. У пацієнтів у групах лікування препаратом Вабісмо в дослідженнях TENAYA і LUCERNE було досягнуто покращення сумарного індексу за NEI VFQ-25 на ≥ 4 бали від вихідного показника на 48 тижні. Таке покращення зберігалось до 112 тижня.
Лікування ДМН
Безпека та ефективність застосування фарицимабу порівняно з анти-VEGF лікуванням вивчались у двох рандомізованих (1:1:1) багатоцентрових подвійно сліпих дослідженнях з трьома групами (YOSEMITE і RHINE) тривалістю 2 роки за участю пацієнтів із ДМН. Пацієнти в трьох досліджуваних групах отримували інтравітреальні ін’єкції фарицимабу по 6 мг кожні 8 тижнів (після 6 щомісячних ін’єкцій на початку лікування), фарицимаб по 6 мг із персоналізованим інтервалом між ін’єкціями максимум до 16 тижнів (після 4 щомісячних ін’єкцій на початку лікування) або афліберцепт по 2 мг кожні 8 тижнів (після 5 щомісячних ін’єкцій на початку лікування).
У групі фарицимабу з подовженим інтервалом між введенням до кожних 16 тижнів дозування препарату виконувалося за стандартизованою схемою лікування з продовженням інтервалів між введеннями доз препарату. Залежно від зміни показника CST за результатами ОКT та/або зміни BCVA, визначеної за показником кількості літер за ETDRS, персоналізований інтервал між ін’єкціями в групі фарицимабу міг бути подовжений на 4 тижні або скорочений на 4 або 8 тижнів під час кожного візиту введення лікарського засобу (див. розділ «Спосіб застосування та дози»).
У дослідження було включено загалом 1891 пацієнта (з яких приблизно 94 % мали цукровий діабет 2 типу), 1622 (85,8 %) пацієнти завершили участь у дослідженні до тижня 100. Загалом 1887 пацієнтів отримали щонайменше одну дозу до тижня 56 (1262 пацієнти отримали препарат Вабісмо). Середній вік [діапазон віку] досліджуваних пацієнтів становив 62,2 року [від 24 до 91 року]. Досліджувана популяція включала пацієнтів, які раніше не отримували анти-VEGF лікування (78 %), і пацієнтів, які раніше отримували анти-VEGF терапію (22 %).
Первинною кінцевою точкою ефективності була середня зміна показника BCVA від вихідного рівня до кінця першого року (середнє 48, 52 і 56 тижнів), визначена за допомогою таблиці літер ETDRS з відстані 4 метри. В обох дослідженнях первинна гіпотеза (не менша ефективність) була підтверджена для обох груп лікування: пацієнти, які отримували препарат Вабісмо кожні 8 тижнів, та пацієнти, які отримували препарат Вабісмо в режимі подовженого інтервалу між введеннями до кожних 16 тижнів, мали порівнянну середню зміну показника BCVA від вихідного рівня, як і пацієнти, які отримували лікування афліберцептом кожні 8 тижнів, через 1 рік, і це покращення зору зберігалось до 2 року.
Після 4 початкових щомісячних введень пацієнти з групи препарату Вабісмо із коригованим інтервалом введень до кожних 16 тижнів могли отримати загалом щонайменше 6 і максимум 21 ін’єкцію до 96 тижня. На 52 тижні у дослідженнях YOSEMITE і RHINE відповідно у 74 % і 71 % пацієнтів у відповідних групах препарату Вабісмо з коригованим режимом дозування кожні 16 тижнів досягнуто інтервалу між введеннями кожні 16 або 12 тижнів (53 % і 51 % – кожні 16 тижнів, 21 % і 20 % – кожні 12 тижнів). Відповідно у 75 % і 84 % цих пацієнтів у дослідженнях YOSEMITE і RHINE зберігався інтервал між введеннями кожні ≥ 12 тижнів без скорочення цього інтервалу менше 12 тижнів до 96 тижня; на 52 тижні 70 % і 82 % пацієнтів, які отримували препарат у режимі кожні 16 тижнів, продовжували отримувати препарат кожні 16 тижнів без зменшення інтервалу до 96 тижня. На 96 тижні в обох дослідженнях у 78 % пацієнтів відповідної групи лікування препаратом Вабісмо із коригованим режимом введення кожні 16 тижнів досягнуто частоти введення кожні 16 або кожні 12 тижнів (60 % і 65 % пацієнтів – кожні 16 тижнів, 18 % і 14 % пацієнтів – кожні 12 тижнів). У 4 % та 6 % пацієнтів у дослідженнях YOSEMITE і RHINE відповідно інтервал було подовжено до 8 тижнів і пацієнти продовжили отримувати препарат з інтервалом введення кожні ≤ 8 тижнів до 96 тижня; 3 % і 5 % отримували препарат з режимом введення кожні 4 тижні до кінця 96 тижня.
Tаблиця 3
Результати щодо ефективності під час візитів для оцінки первинної кінцевої точки у 1a і на 2b роки в дослідженні YOSEMITE
|
Результати щодо ефективності |
YOSEMITE |
|||||
|
1 рік |
2 роки |
|||||
|
Вабісмо кожні 8 тижнів, N = 315 |
Вабісмо з коригованим режимом дозування до кожних 16 тижнів, N = 313 |
Афліберцепт кожні 8 тижнів, N = 312 |
Вабісмо кожні 8 тижнів, N = 262 |
Вабісмо з коригованим режимом дозування до кожних 16 тижнів, N = 270 |
Афліберцепт кожні 8 тижнів, N = 259 |
|
|
Середня зміна від вихідного рівня показника BCVA за результатами визначення кількості літер за ETDRS (97,5 % ДІ в 1 рік і 95 % на 2 рік) |
10,7 (9,4, 12,0) |
11,6 (10,3, 12,9) |
10,9 (9,6, 12,2) |
10,7 (9,4, 12,1) |
10,7 (9,4, 12,1) |
11,4 (10,0, 12,7) |
|
Частка пацієнтів, у яких BCVA збільшилась щонайменше на 15 літер від вихідного рівня (зважена частка за CMH, 95 % ДІ в 1 і на 2 рік) |
29,2 % (23,9 %, 34,5 %) |
35,5 % (30,1 %, 40,9 %) |
31,8 % (26,6 %, 37,0 %) |
37,2 % (31,4 %, 42,9 %) |
38,2 % (32,8 %, 43,7 %) |
37,4 % (31,7 %, 43,0 %) |
|
Частка пацієнтів, які уникнули зменшення BCVA щонайменше на 15 літер від вихідного рівня (зважена частка за CMH, 95 % ДІ в 1 і на 2 рік) |
98,1 % (96,5 %, 99,7 %) |
98,6 % (97,2 %, 100,0 %) |
98,9 % (97,6 %, 100,0 %) |
97,6 % (95,7 %, 99,5 %) |
97,8 % (96,1 %, 99,5 %) |
98,0 % (96,2 %, 99,7 %) |
a Середнє тижнів 48, 52, 56.
b Середнє тижнів 92, 96, 100.
BVCA – найкраща скоригована гострота зору; ETDRS – дослідження ефективності раннього лікування діабетичної ретинопатії; ДІ – довірчий інтервал; CMH – метод Кокрана – Мантеля – Хензеля, статистичний тест, який генерує оцінку зв’язку з бінарним результатом і використовується для оцінки категоріальних змінних.
Примітка: дані щодо CMH-зваженого %, продемонстровані для групи афліберцепту, призначені для порівняння між дозуванням препарату Вабісмо кожні 8 тижнів і афліберцепту, однак відповідний CMH-зважений % для порівняння коригованого режиму препарату Вабісмо з афліберцептом є подібним до зазначеного вище.
ETDRS-DRSS: шкала тяжкості діабетичної ретинопатії за дослідженням ефективності раннього лікування діабетичної ретинопатії (шкала оцінки діабетичної ретинопатії з дослідження ефективності раннього лікування діабетичної ретинопатії).
Tаблиця 4
Частка пацієнтів із покращенням ETDRS-DRSS на ≥ 2 ступеня на 52 і 96 тижнях порівняно з вихідним станом в дослідженні YOSEMITE (оцінювана популяція щодо діабетичної ретинопатії)
|
Результати щодо ефективності |
YOSEMITE |
|||||
|
52 тижні |
96 тижнів |
|||||
|
Вабісмо кожні 8 тижнів, n = 237 |
Вабісмо із коригованим режимом дозування до кожних 16 тижнів, n = 242 |
Афліберцепт кожні 8 тижнів, n = 229 |
Вабісмо кожні 8 тижнів, n = 220 |
Вабісмо із коригованим режимом дозування до кожних 16 тижнів, n = 234 |
Афліберцепт кожні 8 тижнів, n = 221 |
|
|
Частка пацієнтів із покращенням на ≥ 2 ступеня від вихідного рівня за шкалою ETDRS-DRSS (зважена частка за CMH) |
46,0 % |
42,5 % |
35,8 % |
51,4 % |
42,8 % |
42,2 % |
ETDRS-DRSS – шкала тяжкості діабетичної ретинопатії за дослідженням ефективності раннього лікування діабетичної ретинопатії.
ДІ – довірчий інтервал; CMH – метод Кокрана – Мантеля – Хензеля, статистичний тест, який генерує оцінку зв’язку з бінарним результатом і використовується для оцінки категоріальних змінних.
Примітка: дані щодо CMH-зваженого %, продемонстровані для групи афліберцепту, призначені для порівняння між дозуванням препарату Вабісмо кожні 8 тижнів і афліберцепту, однак відповідний CMH-зважений % для порівняння коригованого режиму препарату Вабісмо з афліберцептом є подібним до зазначеного вище.
Tаблиця 5
Результати щодо ефективності під час візитів для оцінки первинної кінцевої точки у 1a і на 2b роки в дослідженні RHINE
|
Результати щодо ефективності |
RHINE |
|||||
|
1 рік |
2 роки |
|||||
|
Вабісмо кожні 8 тижнів, N = 317 |
Вабісмо із коригованим режимом дозування до кожних 16 тижнів, N = 319 |
Афліберцепт кожні 8 тижнів, N = 315 |
Вабісмо кожні 8 тижнів, N = 259 |
Вабісмо із коригованим режимом дозування до кожних 16 тижнів, N = 282 |
Афліберцепт кожні 8 тижнів, N = 254 |
|
|
Середня зміна від вихідного рівня показника BCVA за результатами визначення кількості літер за ETDRS (97,5 % ДІ в 1 рік і 95 % на 2 рік) |
11,8 (10,6, 13,0) |
10,8 (9,6, 11,9) |
10,3 (9,1, 11,4) |
10,9 (9,5, 12,3) |
10,1 (8,7, 11,5) |
9,4 (7,9, 10,8) |
|
Частка пацієнтів, у яких BCVA збільшилась щонайменше на 15 літер від вихідного рівня (зважена частка за CMH, 95 % ДІ в 1 і на 2 рік) |
33,8 % (28,4 %, 39,2 %) |
28,5 % (23,6 %, 33,3 %) |
30,3 % (25,0 %, 35,5 %) |
39,8 % (34,0 %, 45,6 %) |
31,1 % (26,1 %, 36,1 %) |
39,0 % (33,2 %, 44,8 %) |
|
Частка пацієнтів, які уникнули зменшення BCVA щонайменше на 15 літер від вихідного рівня (зважена частка за CMH, 95 % ДІ в 1 і на 2 рік) |
98,9 % (97,6 %, 100,0 %) |
98,7 % (97,4 %, 100,0 %) |
98,6 % (97,2 %, 99,9 %) |
96,6 % (94,4 %, 98,8 %) |
96,8 % (94,8 %, 98,9 %) |
97,6 % (95,7 %, 99,5 %) |
a Середнє тижнів 48, 52, 56.
b Середнє тижнів 92, 96, 100.
BVCA – найкраща скоригована гострота зору; ETDRS – дослідження ефективності раннього лікування діабетичної ретинопатії; ДІ – довірчий інтервал; CMH – метод Кокрана – Мантеля – Хензеля, статистичний тест, який генерує оцінку зв’язку з бінарним результатом і використовується для оцінки категоріальних змінних.
Примітка: дані щодо CMH-зваженого %, продемонстровані для групи афліберцепту, призначені для порівняння між дозуванням препарату Вабісмо кожні 8 тижнів і афліберцепту, однак відповідний CMH-зважений % для порівняння коригованого режиму препарату Вабісмо з афліберцептом є подібним до зазначеного вище.
ETDRS-DRSS – шкала тяжкості діабетичної ретинопатії за дослідженням ефективності раннього лікування діабетичної ретинопатії (шкала оцінки діабетичної ретинопатії з дослідження ефективності раннього лікування діабетичної ретинопатії).
Tаблиця 6
Частка пацієнтів із покращенням ETDRS-DRSS на ≥ 2 ступеня на 52 і 96 тижнях порівняно з вихідним станом в дослідженні RHINE (оцінювана популяція щодо діабетичної ретинопатії)
|
Результати щодо ефективності |
RHINE |
|||||
|
52 тижні |
96 тижнів |
|||||
|
Вабісмо кожні 8 тижнів, n = 237 |
Вабісмо із коригованим режимом дозування до кожних 16 тижнів, n = 242 |
Афліберцепт кожні 8 тижнів, n = 229 |
Вабісмо кожні 8 тижнів, n = 220 |
Вабісмо із коригованим режимом дозування до кожних 16 тижнів, n = 234 |
Афліберцепт кожні 8 тижнів, n = 221 |
|
|
Частка пацієнтів із покращенням на ≥ 2 ступеня за шкалою ETDRS-DRSS від вихідного рівня (зважена частка за CMH) |
44,2 % |
43,7 % |
46,8 % |
53,5 % |
44,3 % |
43,8 % |
ETDRS – дослідження ефективності раннього лікування діабетичної ретинопатії.
ДІ – довірчий інтервал; CMH – метод Кокрана – Мантеля – Хензеля, статистичний тест, який генерує оцінку зв’язку з бінарним результатом і використовується для оцінки категоріальних змінних.
Примітка: дані щодо CMH-зваженого %, продемонстровані для групи афліберцепту, призначені для порівняння між дозуванням препарату Вабісмо кожні 8 тижнів і афліберцепту, однак відповідний CMH-зважений % для порівняння коригованого режиму препарату Вабісмо з афліберцептом є подібним до зазначеного вище.
|
Скоригована середня зміна від вихідного показника |
|
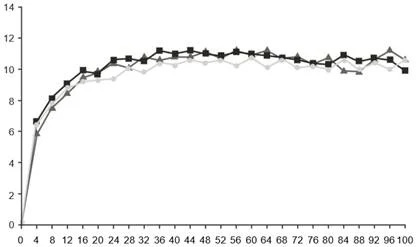
Тижні
 Фарицимаб, 6 мг, кожні 16 тижнів коригованого дозування (N = 632)
Фарицимаб, 6 мг, кожні 16 тижнів коригованого дозування (N = 632)
 Фарицимаб, 6 мг, кожні 8 тижнів (N = 632)
Фарицимаб, 6 мг, кожні 8 тижнів (N = 632)
 Афліберцепт, 2 мг, кожні 8 тижнів (N = 627)
Афліберцепт, 2 мг, кожні 8 тижнів (N = 627)
Рис. 2. Об’єднані дослідження ІII фази ДМН (YOSEMITE і RHINE): графік зміни BCVA від вихідного рівня в досліджуваному оці до тижня 100: метод MMRM (первинна оцінка) (популяція ITT)
Результати щодо ефективності у пацієнтів, які не отримували анти-VEGF лікування до участі в цьому дослідженні, і в усіх інших підгрупах пацієнтів, що підлягали оцінці (зокрема, за віком, статтю, етнічним походженням, вихідним рівнем HbA1c, вихідним показником гостроти зору), в кожному дослідженні збігалися з результатами у відповідній популяції.
Ефект лікування не залежав від глікемічного контролю, і порівнянні результати були досягнуті при лікуванні фарицимабом пацієнтів із покращенням або погіршенням з часом рівня HbA1c на > 0,5 % або залишався в межах 0,5 % від вихідного рівня.
Пацієнти літнього віку
У чотирьох клінічних дослідженнях III фази приблизно 60 % (1149/1929) пацієнтів, рандомізованих для лікування препаратом Вабісмо, були віком ≥ 65 років. Популяційний фармакокінетичний аналіз показав вплив віку на фармакокінетику фарицимабу в органі зору, який був розцінений як клінічно незначущий (див. розділи «Спосіб застосування та дози», «Фармакокінетика»).
Імуногенність
Існує потенціал для розвитку імунної відповіді у пацієнтів, які отримують лікування препаратом Вабісмо (див. розділ «Особливості застосування»).
Після введення препарату Вабісмо до 48 (нВМД) і 100 (ДМН) тижнів антитіла до фарицимабу, які утворилися під час лікування, були виявлені приблизно у 10 % пацієнтів. Клінічне значення антитіл до фарицимабу щодо безпеки дотепер невідоме. У пацієнтів з антитілами до фарицимабу спостерігалася вища частота побічних реакцій у вигляді внутрішньоочного запалення. Однак загальна частота виявлення антитіл до фарицимабу та внутрішньоочного запалення в усій досліджуваній популяції становить приблизно 1 %. Антитіла до фарицимабу не асоціювались із впливом на клінічну ефективність або системну фармакокінетику.
Фармакокінетика.
Абсорбція
Препарат Вабісмо вводиться інтравітреально для здійснення місцевого впливу на око. Клінічні дослідження інших шляхів введення не проводились.
За даними популяційного фармакокінетичного аналізу (включаючи нВМД і ДМН, N = 2246), максимальна концентрація в плазмі крові (Cmax) вільного (незв’язаного з VEGF-A та Ang-2) фарицимабу за розрахунками досягається приблизно через 2 дні після введення. Середня (± стандартне відхилення) Cmax в плазмі крові вільної форми розрахована на рівні 0,23 (0,07) мкг/мл і 0,22 (0,07) мкг/мл відповідно у пацієнтів із нВМД та ДМН/діабетичною ретинопатією (ДР). Після повторних введень середня мінімальна концентрація вільного фарицимабу в плазмі крові прогнозується на рівні 0,002–0,003 мкг/мл при введенні кожні 8 тижнів.
Фарицимаб продемонстрував пропорційну дозі фармакокінетику (на основі Cmax та AUC) в діапазоні доз 0,5–6 мг. Не відмічалося кумуляції фарицимабу в склистому тілі або в плазмі крові після щомісячного введення з огляду на показники експозиції, отримані в популяційній фармакокінетичній моделі.
Розподіл
Немає даних.
Метаболізм
Метаболізм фарицимабу безпосередньо не вивчався. Припускається, що фарицимаб катаболізується на невеликі пептиди та амінокислоти в лізосомах подібно до ендогенних молекул IgG.
Виведення
Профіль концентрація-час фарицимабу в плазмі крові знижувався паралельно з профілем концентрація-час у склистому тілі та внутрішньоочній рідині. Розрахунковий середній період напіввиведення із ока та очевидний системний період напіввиведення фарицимабу становлять по 7,5 дня.
Особливі групи пацієнтів
Порушення функції печінки
Формальне дослідження фармакокінетики у пацієнтів із порушенням функції печінки не проводилось.
Порушення функції нирок
Формальне дослідження фармакокінетики у пацієнтів із порушенням функції нирок не проводилось.
Фармакокінетичний аналіз даних пацієнтів в усіх клінічних дослідженнях, включаючи 857 пацієнтів із легким, 532 пацієнти із помірним та 37 пацієнтів із тяжким порушенням функції нирок, не виявив відмінності в системній фармакокінетиці фарицимабу після інтравітреального введення препарату Вабісмо.
Пацієнти літнього віку
У чотирьох клінічних дослідженнях III фази приблизно 60 % (1149/1929) пацієнтів, рандомізованих для лікування препаратом Вабісмо, були віком ≥ 65 років. Популяційний фармакокінетичний аналіз показав вплив віку на фармакокінетику фарицимабу в органах зору, який, однак, був розцінений як клінічно незначущий.
Інші демографічні фактори
Популяційний фармакокінетичний аналіз показав вплив маси тіла на фармакокінетику фарицимабу в оці, а також на системну фармакокінетику фарицимабу. Цей ефект був розцінений як клінічно незначущий; з огляду на це корекція дози не потрібна.
Популяційно-кінетичний аналіз свідчить про відсутність будь-якого впливу етнічного походження або статі пацієнта на системну фармакокінетику препарату Вабісмо.
Показания Вабисмо раствор д/ин. 120 мг/мл по 0.24 мл (28.8 мг) №1 во флак. с иголк.
Лікування неоваскулярної (ексудативної) вікової макулярної дегенерації (нВМД).
Лікування діабетичного макулярного набряку (ДМН).
Противопоказания Вабисмо раствор д/ин. 120 мг/мл по 0.24 мл (28.8 мг) №1 во флак. с иголк.
Інфекції очей або періокулярні інфекції.
Активне внутрішньоочне запалення.
Встановлена гіперчутливість до фарицимабу або до будь-якої допоміжної речовини лікарського засобу. Реакції гіперчутливості можуть проявлятися висипанням, свербежем, кропив’янкою, еритемою або тяжким внутрішньоочним запаленням.
Взаємодія з іншими лікарськими засобами та інші види взаємодії
Дослідження взаємодії інших лікарських засобів з препаратом Вабісмо не проводились.
Особливості щодо застосування
Реакції, пов’язані з інтравітреальною ін’єкцією
Інтравітреальні ін’єкції, у тому числі препарату Вабісмо, асоціювалися з ендофтальмітом, внутрішньоочним запаленням, регматогенним відшаруванням сітківки, розривом сітківки і ятрогенною травматичною катарактою. При введенні препарату Вабісмо необхідно завжди дотримуватися належної асептичної техніки ін’єкції. Пацієнтів потрібно проінструктувати про необхідність негайно повідомляти про будь-які симптоми, такі як біль, втрата зору, фотофобія, нечіткість зору, плаваюче помутніння або почервоніння, що свідчать про ендофтальміт або будь-яке із зазначених вище явищ, з метою забезпечення своєчасного та належного лікування.
Tранзиторне підвищення внутрішньоочного тиску (ВОТ) спостерігалося протягом 60 хвилин після інтравітеральної ін’єкції, у тому числі при введенні препарату Вабісмо. Особливі запобіжні заходи необхідні для пацієнтів із погано контрольованою глаукомою (не вводити препарат Вабісмо при ВОТ ≥ 30 мм рт. ст.). В усіх випадках як ВОТ, так і перфузію диска зорового нерва та/або гостроту зору необхідно контролювати відповідним чином та лікувати у разі необхідності.
Системні ефекти
Повідомлялося про системні небажані явища, у тому числі про артеріальні тромбоемболії. Існує теоретичний ризик, що ці явища можуть бути пов’язані з пригніченням фактора росту ендотелію судин (VEGF).
Імуногенність
Діюча речовина препарату Вабісмо є терапевтичним білком. Через це можливе виникнення імунологічної реакції на препарат Вабісмо. Пацієнтів потрібно проінструктувати про необхідність повідомляти про будь-які ознаки або симптоми внутрішньоочного запалення, такі як втрата зору, біль в очах, підвищення чутливості до світла, плаваючі помутніння або посилення почервоніння очей, які можуть бути клінічними ознаками, пов’язаними з гіперчутливістю.
Двостороннє лікування
Безпека та ефективність препарату Вабісмо при введенні в обидва ока не вивчались.
Супутнє застосування інших анти-VEGF лікарських засобів
Немає даних щодо супутнього застосування препарату Вабісмо з анти-VEGF лікарськими засобами в одне око.
Призупинення лікування
Лікування слід початково призупинити пацієнтам із:
- регматогенним відшаруванням сітківки, перфорацією сітківки 3 або 4 ступеня тяжкості, розривом сітківки; лікування не слід відновлювати, поки не досягнуто загоєння належного рівня;
- пов’язаним з лікуванням зниженням найкращої скоригованої гостроти зору (BCVA) на ≥ 30 літер порівняно з останньою оцінкою гостроти зору; лікування не слід відновлювати раніше наступного запланованого введення препарату;
- виконаним або запланованим внутрішньоочним хірургічним втручанням протягом попередніх або наступних 28 днів; лікування не слід відновлювати раніше наступного запланованого введення препарату.
Розрив пігментного епітелію сітківки
Фактори ризику, що асоціюються із виникненням розриву пігментного епітелію сітківки після анти-VEGF терапії з приводу нВМД, включають поширене та/або високе відшарування пігментного епітелію. Тому при застосуванні пацієнтам з цими факторами ризику виникнення розриву пігментного епітелію сітківки слід дотримуватись обережності на початку терапії препаратом Вабісмо.
Популяції з обмеженими даними
Існує лише обмежений досвід лікування пацієнтів із ДМН із рівнем HbA1c більше 10 %, пацієнтів із проліферативною діабетичною ретинопатією з високим ризиком або пацієнтів із нВМД та ДМН з активними системними інфекціями. Також немає досвіду лікування препаратом Вабісмо хворих на цукровий діабет із неконтрольованою артеріальною гіпертензією. Лікарю необхідно враховувати відсутність цієї інформації при лікуванні таких пацієнтів.
Зловживання лікарськими засобами та залежність
Відсутні дані щодо можливості розвитку зловживання та залежності при застосуванні препарату Вабісмо.
Інша інформація
Препарат Вабісмо, розчин для ін’єкцій для інтравітреального застосування, містить менше 1 ммоль (23 мг)/дозу натрію, тобто практично вільний від натрію.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
Препарат Вабісмо має незначний вплив на здатність керувати транспортними засобами та працювати з іншими механізмами через можливі тимчасові порушення зору після інтравітреальної ін’єкції і пов’язаного з цим обстеження ока. Пацієнтам не слід керувати транспортними засобами та працювати з іншими механізмами до достатнього відновлення функції зору.
Несумісність
За відсутності досліджень несумісності цей лікарський засіб не слід змішувати з іншими лікарськими засобами.
Способ применения и дозы Вабисмо раствор д/ин. 120 мг/мл по 0.24 мл (28.8 мг) №1 во флак. с иголк.
Загальна інформація
Лише для інтравітреальної ін’єкції. Препарат Вабісмо повинен вводити кваліфікований лікар, який має досвід виконання інтравітреальних ін’єкцій. Препарат з одного флакона слід використовувати лише для лікування одного ока.
Однодозовий скляний флакон з препаратом Вабісмо містить 28,8 мг фарицимабу у 0,24 мл розчину. Це забезпечує необхідну кількість розчину для ін’єкції 0,05 мл, що містить 6 мг фарицимабу як однократну дозу.
Неоваскулярна (ексудативна) вікова макулярна дегенерація (нВМД)
Рекомендована доза препарату Вабісмо становить 6 мг (0,05 мл) шляхом інтравітреальної ін’єкції кожні 4 тижні (приблизно кожні 28 ± 7 днів або один раз на місяць) для перших 4 введень. Надалі графік лікування можна індивідуалізувати з подовженням інтервалів між введеннями препарату. На підставі оцінки лікарем товщини центральної частини сітківки пацієнта (товщина центрального підполя, CST) та/або показника гостроти зору інтервал між введеннями можна подовжити максимум до кожних 16 тижнів (4 місяці). Інтервал між введеннями необхідно відповідно зменшити у разі погіршення показника CST та/або показника гостроти зору (див. розділ «Фармакодинаміка»).
Деякі пацієнти можуть потребувати введення з частотою кожні 4 тижні (приблизно кожні 28 ± 7 днів або один раз на місяць).
Mоніторинг між візитами введення препарату слід планувати залежно від стану пацієнта та на розсуд лікаря.
Діабетичний макулярний набряк (ДМН)
Рекомендована доза препарату Вабісмо становить 6 мг (0,05 мл) шляхом інтравітреальної ін’єкції кожні 4 тижні (приблизно кожні 28 ± 7 днів або один раз на місяць) для перших 4 введень. Надалі графік лікування можна індивідуалізувати за допомогою режиму лікування з подовженням інтервалів між введеннями препарату. На основі оцінки лікарем CST та/або гостроти зору окремого пацієнта інтервал між введеннями можна подовжити максимум до кожних 16 тижнів (4 місяці). Інтервал між введеннями необхідно відповідним чином зменшити у разі погіршення CST та/або гостроти зору (див. розділ «Фармакодинаміка»).
Mоніторинг між візитами введення препарату слід планувати залежно від стану пацієнта та на розсуд лікаря, однак немає вимоги щомісячного моніторингу між ін’єкціями.
З метою забезпечення відстеження біологічних лікарських засобів рекомендується документувати торгівельну назву і номер серії при кожному введенні лікарського засобу.
Тривалість лікування
Препарат Вабісмо призначений для довгострокового лікування.
Корекція дози після виникнення небажаних ефектів/взаємодій
Зміна дози препарату Вабісмо не рекомендується.
Відкладене введення
Якщо ін’єкцію відкладено або пропущено, пацієнту необхідно повернутись на обстеження лікаря під час наступного доступного візиту і продовжувати введення препарату залежно від рішення лікаря.
Якщо результати візуального та/або анатомічного обстеження свідчать, що пацієнт не отримує користі від продовження лікування, лікування препаратом Вабісмо слід припинити.
Особливі групи пацієнтів
Порушення функції печінки
Спеціальні дослідження препарату Вабісмо за участю пацієнтів із порушенням функції печінки не проводились (див. розділ «Фармакокінетика»).
Однак корекція дози не потрібна пацієнтам із порушенням функції печінки.
Порушення функції нирок
Спеціальні дослідження препарату Вабісмо за участю пацієнтів із порушенням функції нирок не проводились (див. розділ «Фармакокінетика»).
Однак корекція дози не потрібна пацієнтам із захворюваннями нирок.
Пацієнти літнього віку
У чотирьох дослідженнях III фази приблизно 60 % (1149/1929) пацієнтів, рандомізованих для отримання лікування препаратом Вабісмо, були віком ≥ 65 років. Популяційний фармакокінетичний аналіз продемонстрував вплив віку на фармакокінетику фарицимабу в органах зору. Однак ефект був оцінений як клінічно не значущий. У цих дослідженнях не було виявлено суттєвої відмінності в ефективності чи безпеці застосування фарицимабу зі збільшенням віку пацієнта. Корекція дози пацієнтам віком ≥ 65 років не потрібна (див. розділ «Фармакокінетика»).
Діти
Безпека та ефективність застосування препарату Вабісмо дітям не встановлені.
Особливі групи пацієнтів
Спеціальна корекція дози не потрібна для будь-якої з досліджуваних популяцій пацієнтів (зокрема, за віком (літні пацієнти), за статтю та расою).
Інструкції щодо застосування
Перед введенням препарат Вабісмо необхідно оглянути візуально щодо наявності сторонніх часток і зміни забарвлення.
Безпосередньо після інтравітреальної ін’єкції за станом пацієнтів необхідно спостерігати щодо підвищення внутрішньоочного тиску. Належний моніторинг може складатись із перевірки перфузії диска зорового нерва або тонометрії. Стерильне обладнання для парацентезу повинно бути доступним у разі необхідності.
Після інтравітреальної ін’єкції пацієнтів слід проінструктувати про необхідність негайно повідомляти про будь-які симптоми, що свідчать про ендофтальміт (наприклад, втрата зору, біль в очах, почервоніння очей, фотофобія, нечіткість зору).
Приготування препарату до введення
Препарат Вабісмо є стерильним прозорим або опалесцентним розчином, що не містить консервантів, безбарвним або коричнювато-жовтого кольору.
Препарат Вабісмо необхідно оглянути візуально після виймання з холодильника і перед введенням.
У разі наявності сторонніх часток, помутніння або зміни забарвлення флакон використовувати не можна.
Вміст флакона і голка з фільтром для перенесення є стерильними і призначені лише для однократного використання. Не застосовувати препарат, якщо упаковка, флакон та/або голка з фільтром для перенесення пошкоджені або їхній термін придатності минув.
Під час приготування до інтравітреальної ін’єкції необхідно дотримуватися правил асептики.
Інструкції щодо застосування препарату
Наведена нижче інформація призначена лише для медичних працівників.
Перед початком:
- Уважно прочитати всі інструкції перед застосуванням препарату Вабісмо.
- Упаковка препарату Вабісмо включає скляний флакон з лікарським засобом і голку з фільтром для перенесення. Скляний флакон містить лише однократну дозу. Голка з фільтром призначена лише для однократного використання.
- Препарат Вабісмо слід зберігати в холодильнику при температурі від 2 до 8 °C. Не заморожувати. Не струшувати.
- Перед введенням дати можливість препарату Вабісмо досягти кімнатної температури (від 20 до 25 °C). Зберігати флакон в оригінальній картонній упаковці з метою захисту від світла.
- Флакон препарату Вабісмо можна зберігати при кімнатній температурі протягом 24 годин.
- Перед введенням флакон з препаратом Вабісмо необхідно оглянути візуально. Препарат Вабісмо є прозорим або опалесцентним розчином, безбарвним або коричнювато-жовтого кольору.
Не використовувати препарат, якщо візуалізуються сторонні частки, помутніння або зміна забарвлення.
Не використовувати препарат, якщо упаковка, флакон та/або голка з фільтром для перенесення відкриті, пошкоджені або термін придатності минув (див. рисунок A).
- Під час приготування до інтравітреальної ін’єкції необхідно дотримуватись правил асептики.
Рисунок А
Інструкції щодо використання флакона:
1. Зібрати такі засоби:
- Один флакон препарату Вабісмо (в картонній коробці).
- Одна стерильна голка з фільтром для перенесення з тупим кінцем, 5 мікрон, розміром 18G × 11/2 дюйма або приблизно 1,2 мм × 40 мм (в картонній коробці).
- Один стерильний шприц Luer lock об’ємом 1 мл із позначкою дози 0,05 мл (не додається).
- Одна стерильна голка для ін’єкцій розміром 30G × 1/2 дюйма (не додається).
- Слід зазначити, що голка для ін’єкцій 30G рекомендується з метою уникнення збільшення сили введення, що може бути необхідним при використанні голок меншого діаметру.
- Спиртовий тампон (не додається).
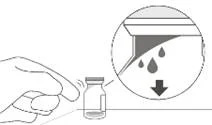
2. Щоб вся рідина осіла на дні флакона, поставити флакон вертикально на плоску поверхню (приблизно на 1 хвилину) після вилучення з упаковки, щоб вся рідина осіла на дні флакона (див. рисунок B). Обережно постукати по флакону пальцем (див. рисунок C), оскільки рідина може прилипнути до верху флакона.
|
|
|
|
Рисунок В |
Рисунок С |
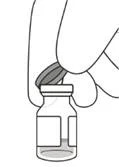
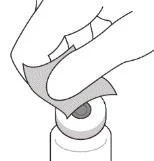
3. Зняти з флакона ковпачок (див. рисунок D) і протерти перегородку флакона спиртовим тампоном (див. рисунок E).
|
|
|
|
Рисунок D |
Рисунок Е |
4. Дотримуючись правил асептики, міцно прикріпити додану голку з фільтром для перенесення 18G × 11/2 дюйма до шприца Luer Lock об’ємом 1 мл (див. рисунок F).
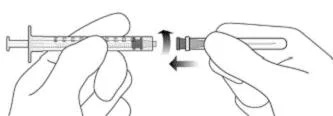
Рисунок F
5. Дотримуючись правил асептики, ввести голку з фільтром для перенесення в центр перегородки флакона (див. рисунок G), втиснути її до кінця у флакон, потім злегка нахилити флакон так, щоб голка торкнулася нижнього краю флакона (див. рисунок H).
|
|
|
|
Рисунок G |
Рисунок Н |
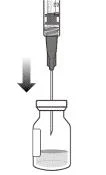
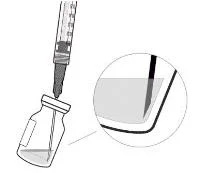
6. Тримати флакон злегка нахиленим і повільно витягнути всю рідину з флакона (див. рисунок I). Тримати кінець голки з фільтром для перенесення зануреним у рідину, щоб уникнути потрапляння повітря.
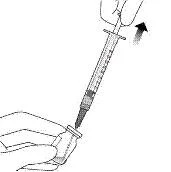
Рисунок I
7. Переконатися, що шток поршня достатньо відтягнутий назад під час спорожнення флакона, щоб повністю спорожнити голку з фільтром для перенесення (див. рисунок I).
8. Від’єднати голку з фільтром для перенесення від шприца та утилізувати її відповідно до місцевих вимог.
Не використовувати голку з фільтром для перенесення для інтравітреальної ін’єкції.
9. Дотримуючись правил асептики, міцно прикріпити голку для ін’єкцій 30G × 11/2 дюйма до шприца Luer Lock (див. рисунок J).
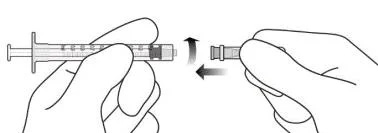
Рисунок J
10. Обережно зняти пластиковий ковпачок голки з голки, потягнувши його.
11. Щоб перевірити наявність бульбашок повітря, тримати шприц голкою вгору. Якщо є бульбашки повітря, обережно постукати пальцем по шприцу, поки бульбашки не піднімуться наверх (див. рисунок K).
12. Обережно видалити повітря зі шприца та голки та повільно натиснути на поршень, щоб сумістити край гумового ущільнювача поршня з позначкою дози 0,05 мл. Тепер шприц готовий до ін’єкції (див. рисунок L). Переконатися, що ін’єкція виконується негайно після приготування дози.
|
|
|
|
Рисунок К |
Рисунок L |
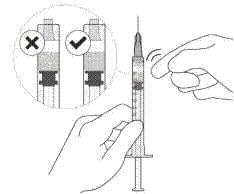
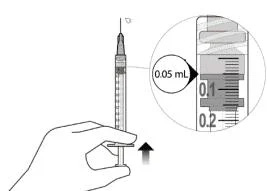
13. Вводити повільно, поки гумовий ущільнювач поршня не досягне передньої частини шприца, щоб ввести об’єм 0,05 мл. Підтвердити введення повної дози, перевіривши, що гумовий ущільнювач досяг передньої частини шприца після ін’єкції.
Будь-який невикористаний лікарський засіб або відходи необхідно утилізувати відповідно до місцевих вимог.
Діти
Безпека та ефективність застосування препарату Вабісмо дітям не встановлені.
Побочные реакции Вабисмо раствор д/ин. 120 мг/мл по 0.24 мл (28.8 мг) №1 во флак. с иголк.
Резюме профілю безпеки
Усі дані з безпеки були отримані у дослідженнях III фази з активним контролем (афліберцепт).
Загалом 3213 пацієнтів увійшли до популяції для оцінки безпеки в чотирьох клінічних дослідженнях III фази тривалістю два роки (1926 пацієнтів, які отримували лікування препаратом Вабісмо: 664 пацієнти із нВМД і 1262 пацієнти із ДМН). Найбільш серйозними побічними реакціями були катаракта (1,0 %), увеїт (0,6 %), ендофтальміт (0,5 %), вітрит (0,3 %), розриви сітківки (0,2 %), регматогенне відшарування сітківки (0,1 %) і травматична катаракта (< 0,1 %).
Найбільш частими побічними реакціями, зареєстрованими у пацієнтів, які отримували лікування препаратом Вабісмо, були катаракта (13 %), кон’юнктивальний крововилив (8 %),
відшарування сітківки (5 %), підвищення ВОТ (4 %), плаваючі помутніння склистого тіла (4 %), біль в очах (3 %) і розрив пігментного епітелію сітківки (лише у пацієнтів із нВМД) (3 %).
Описані нижче дані з безпеки включають усі побічні реакції із сукупних даних чотирьох клінічних досліджень III фази за участю пацієнтів із нВМД та ДМН з обґрунтованою можливістю причинно-наслідкового зв’язку з процедурою ін’єкції або лікарським засобом. Побічні реакції наведено відповідно до класу системи органів за MedDRA і такими категоріями частоти: дуже часто (≥ 1/10), часто (≥ 1/100–< 1/10), нечасто (≥ 1/1 000–< 1/100), рідко (≥ 1/10 000–< 1/1000).
Tаблиця 7
Резюме побічних реакцій у пацієнтів, які отримували лікування препаратом Вабісмо в клінічних дослідженнях III фази
|
Побічні реакції |
Вабісмо, N = 1 926 |
Категорія частоти |
|
Розлади з боку органів зору |
||
|
Катаракта |
12,7 % |
Дуже часто |
|
Кон’юнктивальний крововилив |
8,0 % |
Часто |
|
Відшарування сітківки |
4,8 % |
Часто |
|
Підвищення внутрішньоочного тиску |
4,2 % |
Часто |
|
Плаваючі помутніння склистого тіла |
4,1 % |
Часто |
|
Розрив пігментного епітелію сітківки (лише нВМД) |
2,9 % |
Часто |
|
Біль в очах |
3,0 % |
Часто |
|
Збільшення сльозотечі |
1,1 % |
Часто |
|
Ерозія рогівки |
1,1 % |
Часто |
|
Подразнення очей |
1,0 % |
Часто |
|
Дискомфорт в очах |
0,9 % |
Нечасто |
|
Свербіж в очах |
0,9 % |
Нечасто |
|
Гіперемія очей |
0,8 % |
Нечасто |
|
Нечіткість зору |
0,8 % |
Нечасто |
|
Зниження гостроти зору |
0,7 % |
Нечасто |
|
Ірит |
0,7 % |
Нечасто |
|
Увеїт |
0,6 % |
Нечасто |
|
Ендофтальміт |
0,5 % |
Нечасто |
|
Відчуття стороннього тіла |
0,5 % |
Нечасто |
|
Крововилив у склисте тіло |
0,5 % |
Нечасто |
|
Іридоцикліт |
0,4 % |
Нечасто |
|
Вітрит |
0,3 % |
Нечасто |
|
Гіперемія кон’юнктиви |
0,3 % |
Нечасто |
|
Біль під час проведення процедури |
0,2 % |
Нечасто |
|
Розрив сітківки |
0,2 % |
Нечасто |
|
Регматогенне відшарування сітківки |
0,1 % |
Нечасто |
|
Транзиторне зниження гостроти зору |
< 0,1 % |
Рідко |
|
Травматична катаракта |
< 0,1 % |
Рідко |
Опис окремих побічних реакцій та додаткова інформація
Після інтравітреального введення інгібіторів VEGF існує теоретичний ризик артеріальної тромбоемболії, у тому числі інсульту та інфаркту міокарда. Низька частота артеріальної тромбоемболії спостерігалась у клінічних дослідженнях препарату Вабісмо у пацієнтів із нВМД та ДМН. Не спостерігалося помітної відмінності між групами, які отримували лікування препаратом Вабісмо і препаратом порівняння, при різних показаннях.
Передозировка Вабисмо раствор д/ин. 120 мг/мл по 0.24 мл (28.8 мг) №1 во флак. с иголк.
Дози, що перевищують рекомендовані, не вивчались. Передозування внаслідок перевищення рекомендованого об’єму ін’єкції може призвести до підвищення внутрішньоочного тиску.
У разі передозування необхідно контролювати ВОТ і, якщо лікар вважає доцільним, необхідно розпочати відповідне лікування.
Применение в период беременности или кормления грудью Вабисмо раствор д/ин. 120 мг/мл по 0.24 мл (28.8 мг) №1 во флак. с иголк.
Жінки репродуктивного віку
Жінкам репродуктивного віку необхідно використовувати ефективну контрацепцію під час лікування препаратом Вабісмо і протягом щонайменше 3 місяців після введення останньої дози препарату Вабісмо.
Вагітність
Відсутні дані щодо застосування препарату Вабісмо вагітним жінкам.
Не спостерігалося побічних ефектів у дослідженні на вагітних яванських макаках.
Було показано, що пригнічення VEGF спричиняє вади розвитку, резорбцію ембріона/плода і зниження маси тіла плода. Також було показано, що пригнічення VEGF впливає на розвиток фолікулів, функцію жовтого тіла та фертильність. Немає спеціальних досліджень впливу пригнічення Ang-2 на вагітність. З огляду на доклінічні дані пригнічення Аng-2 може призвести до ефектів, порівнянних із такими при пригніченні VEGF. Системна експозиція після введення препарату Вабісмо в орган зору є дуже низькою.
Невідомо, чи фарицимаб проникає через плаценту або спричиняє шкоду плоду при застосуванні вагітним жінкам. Зважаючи на механізм дії інгібіторів VEGF та Ang-2, існує потенційний ризик для репродуктивного потенціалу жінок і для ембріофетального розвитку. Хоча системна експозиція після введення в орган зору є дуже низькою, фарицимаб не слід застосовувати під час вагітності, за винятком випадків, коли потреба у лікуванні зумовлена клінічним станом жінки.
Пологи
Безпека застосування препарату Вабісмо під час пологів не встановлена.
Годування груддю
Невідомо, чи екскретується препарат Вабісмо в грудне молоко людини. Не проводилися дослідження впливу Вабісмо на продукування молока або наявності препарату в грудному молоці. Оскільки багато лікарських засобів екскретуються в грудне молоко людини із потенціалом до абсорбції і шкоди для росту і розвитку немовляти, слід дотримуватись обережності при застосуванні препарату Вабісмо жінкам, які годують груддю. Необхідно зіставити користь годування груддю для розвитку і здоров’я дитини із клінічною потребою в лікуванні матері препаратом Вабісмо та будь-яким потенційним небажаним впливом препарату Вабісмо на дитину, яка отримує грудне молоко.
Фертильність
Дослідження впливу на репродуктивну функцію або фертильність не проводились. Не спостерігався вплив на репродуктивні органи в 6-місячному дослідженні на яванських макаках при застосуванні фарицимабу в дозах до 3 мг/око (експозиція, що у 8–10 разів перевищує клінічну експозицію, на підставі показника AUC). Було показано, що пригнічення VEGF не впливає на розвиток фолікулів, функцію жовтого тіла та фертильність. Зважаючи на механізм дії інгібіторів VEGF та Ang-2, існує потенційний ризик для репродуктивного потенціалу жінок і ембріофетального розвитку, однак цей ризик вважається низьким через низьку системну експозицію після введення препарату в орган зору.
Условия хранения Вабисмо раствор д/ин. 120 мг/мл по 0.24 мл (28.8 мг) №1 во флак. с иголк.
Термін придатності
30 місяців.
Умови зберігання
Зберігати при температурі від 2 до 8 ºС в оригінальній упаковці з метою захисту від світла.
Не заморожувати. Не струшувати. Зберігати у недоступному для дітей місці.
Перед застосуванням невідкритий флакон препарату Вабісмо можна зберігати при кімнатній температурі (від 20 до 25 °C) протягом 24 годин.
Необхідно забезпечити негайне виконання ін’єкції після приготування дози.
Упаковка
Флакон
Флакон об’ємом 2 мл з безбарвного боросилікатного скла класу І, укупорений гумовою пробкою сірого кольору із бутилкаучуку діаметром 13 мм, ламінованою фторкаучуком, та обтиснутий алюмінієвим ковпачком діаметром 13 мм з пластиковим диском типу «flip-off».
Голка з фільтром
Голка із нержавіючої сталі 18 G з фільтром 5 мкм із акрилового сополімеру з поліпропіленовим ковпачком.
По 1 флакону у комплекті з голкою з фільтром, упакованою в блістер у картонній коробці
Категорія відпуску
За рецептом.
Регистрационные данные Вабисмо раствор д/ин. 120 мг/мл по 0.24 мл (28.8 мг) №1 во флак. с иголк.
Производитель: Ф. Хоффманн-Ля Рош Лтд. (Швейцарія)
Фарм. группа: Средства, применяемые при сосудистых заболеваниях глаз. Антинеоваскуляризацийные средства.
Регистрация: UA/20151/01/01 від 25/08/2023 наказ №794 від 07/05/2024
МНН: Фарицимаб
Код АТХ:
(S) Лекарственные средства для лечения заболеваний органов чувств
(S01) Препараты для лечения заболеваний глаз (офтальмологические препараты)
(S01L) Средства для применения при сосудистых глазных заболеваниях
(S01LA) Протинеоваскуляризационные средства
(S01LA09) Фарицимаб
Дата обновления информации: 14.03.2025 г.
© likiteka 2025
- Айлия р-р д/ин. 40 мг/мл шприц 0,165 мл №1, Байєр АГ (Німеччина)
- Айлия р-р д/ин. 40 мг/мл фл. 0,278 мл, с иглой фильтровальной G18 №1, Байєр АГ (Німеччина)
- Луцентис р-р д/ин. 10 мг/мл фл. 0,23 мл, + шприц, 2 иглы №1, Новартіс Фарма АГ, Швейцарія
- Макуген р-р д/ин. 0,3 мг шприц 90 мкл №1, Пфайзер Ейч. Сі. Пі. Корпорейшн, США
- Визкью раствор д/ин. 120 мг/мл по 0.23 мл №1 во флак. с иголк., Алкон - Куврьор для "Алкон Лабораторіз, Інк", Бельгія/США